Studies at the Intersection of Ribosome Function and Cellular Homeostasis
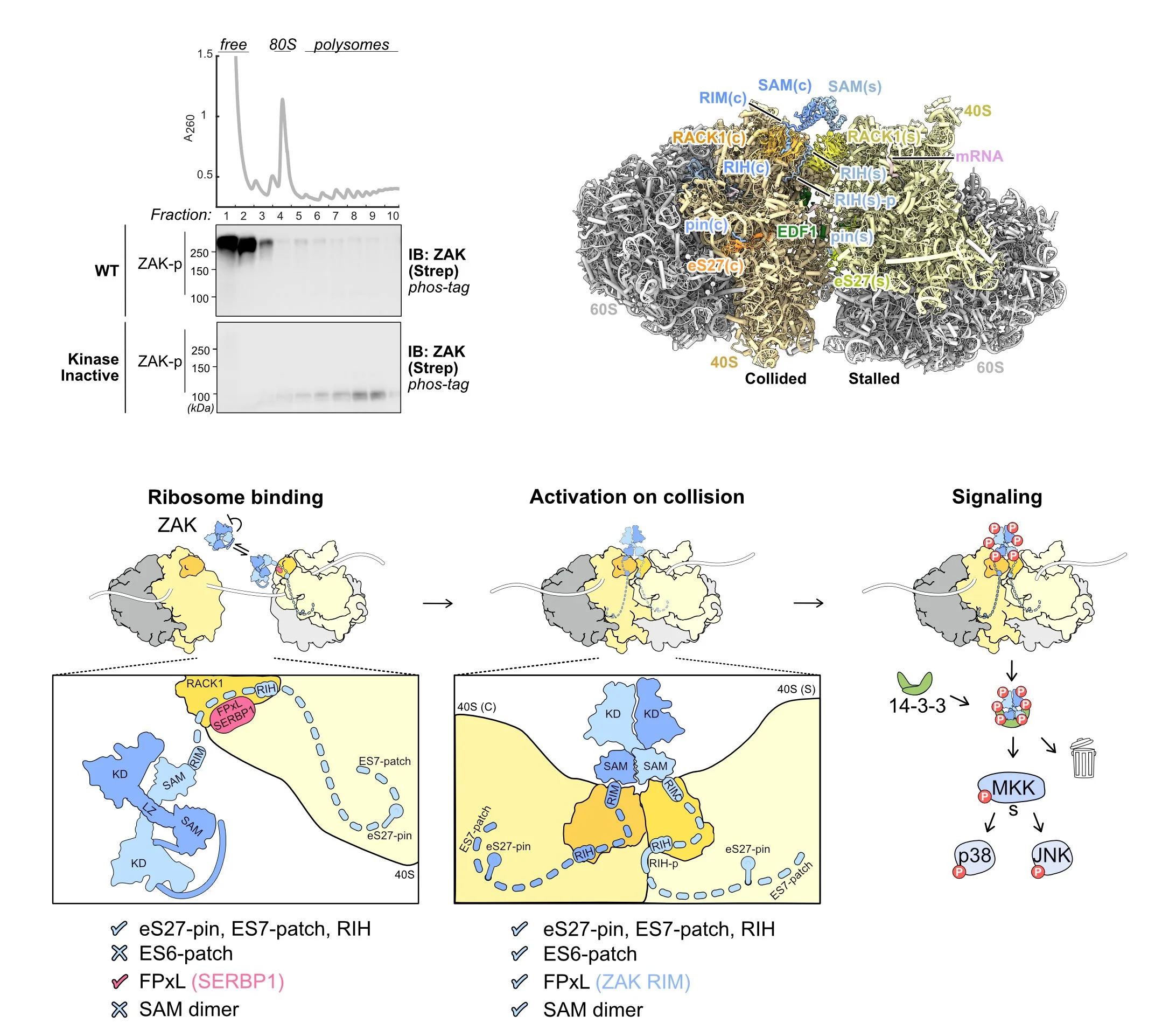
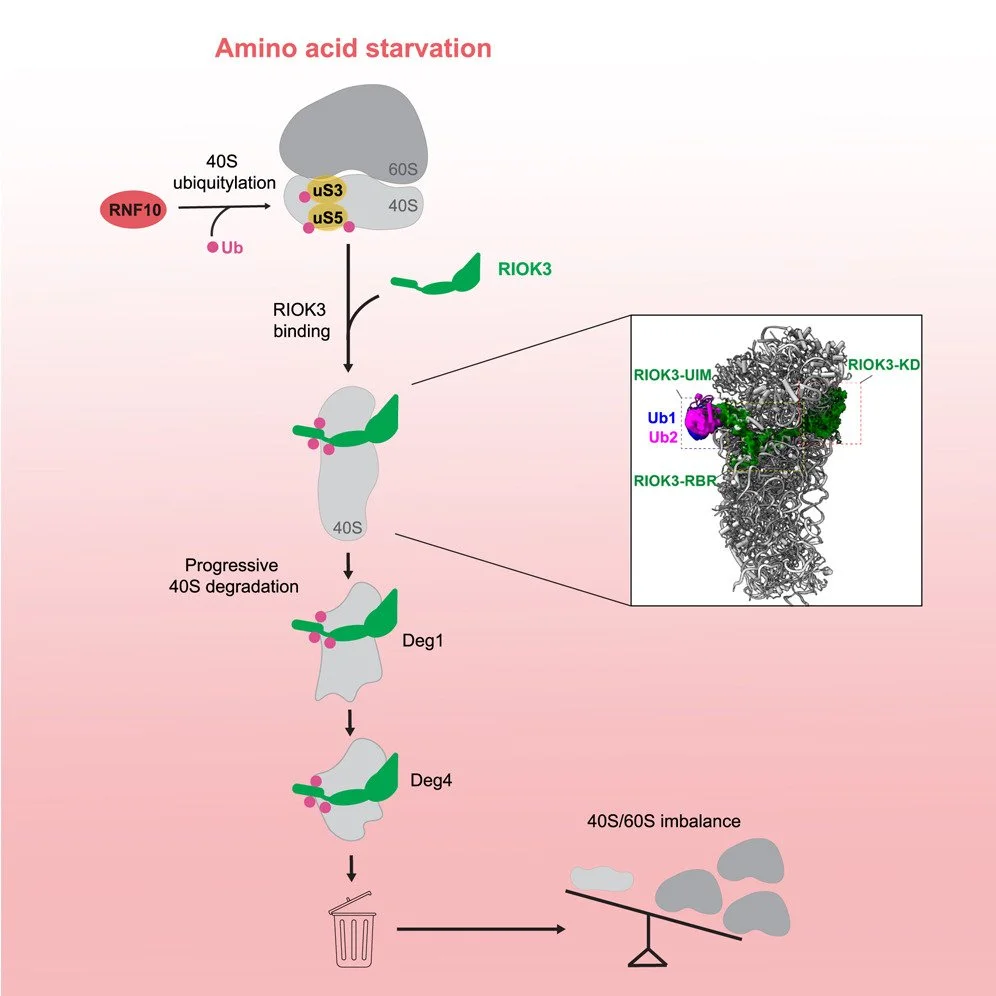
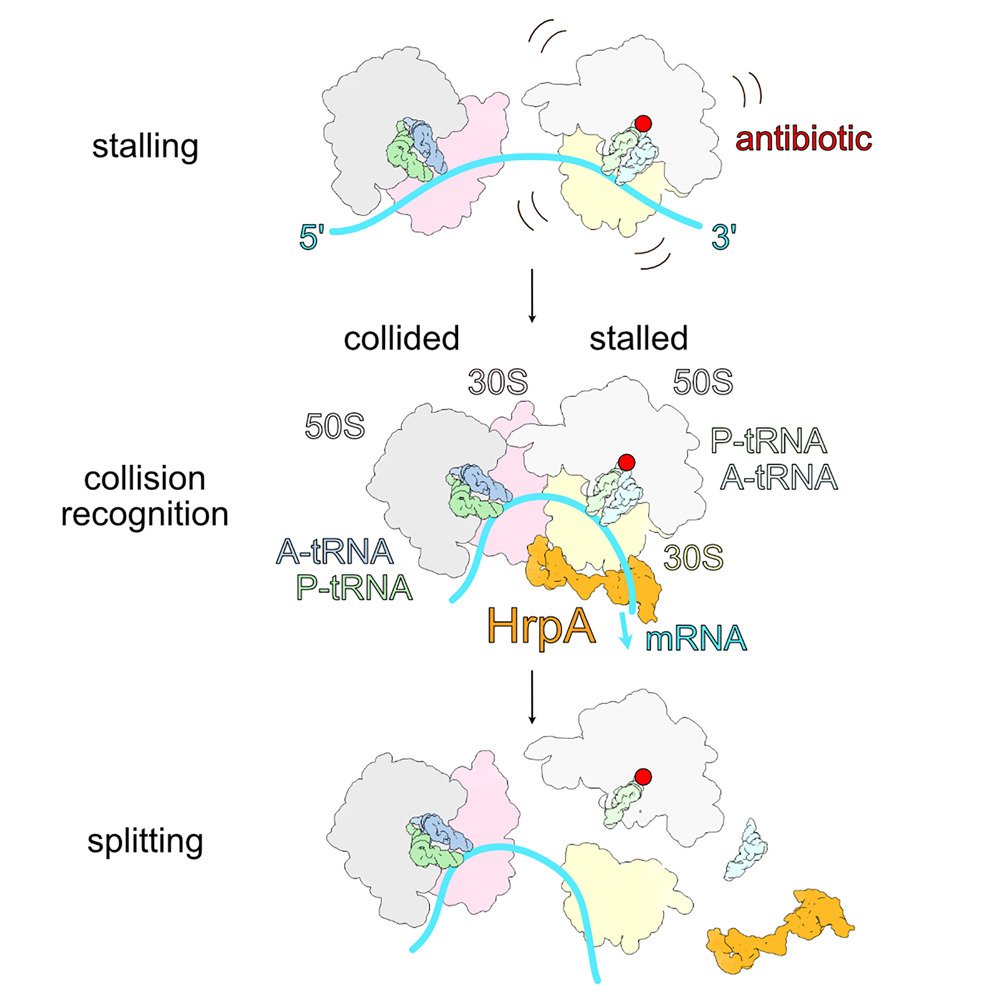
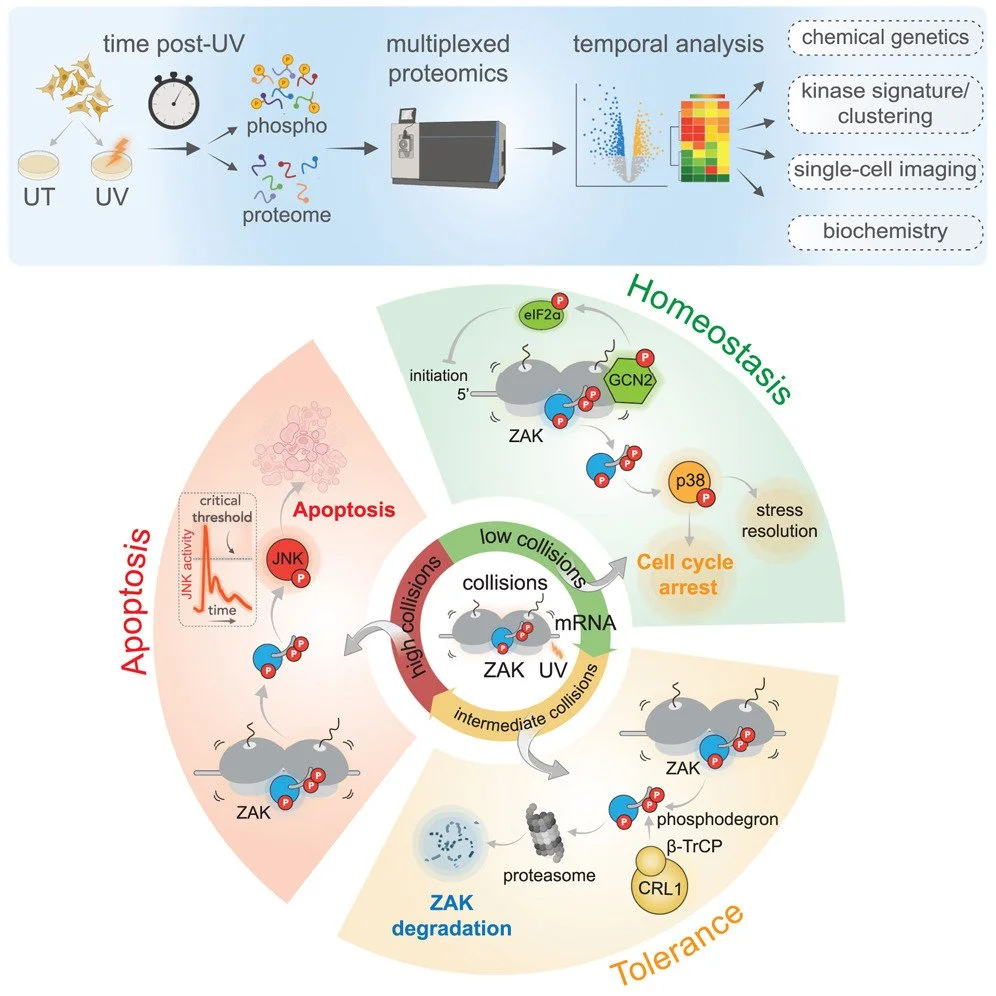
We are broadly interested in the role of ribosomes in cells in translating mRNAs into functional polypeptides and in the regulation of translation in responding to signals from the outside world. Our most recent work has focused on ribosome-mediated quality control and the many factors implicated in this process. We have used genetic and proteomic screens in yeast, mammals and bacteria to identify novel factors involved in these pathways. Once we identify factors of interest, we use a broad range of approaches to characterize the role of these factors in the process with a strong emphasis on molecular mechanism. We have shown that many of these factors specifically recognize collided ribosomes, thus revealing a unifying principle for how cells must assess overall translational distress. We routinely collaborate with cryoEM labs to leverage structural information in our studies and recently published the first atomic resolution structure of a quality control factor (EDF1/Mbf1) bound to colliding ribosomes.
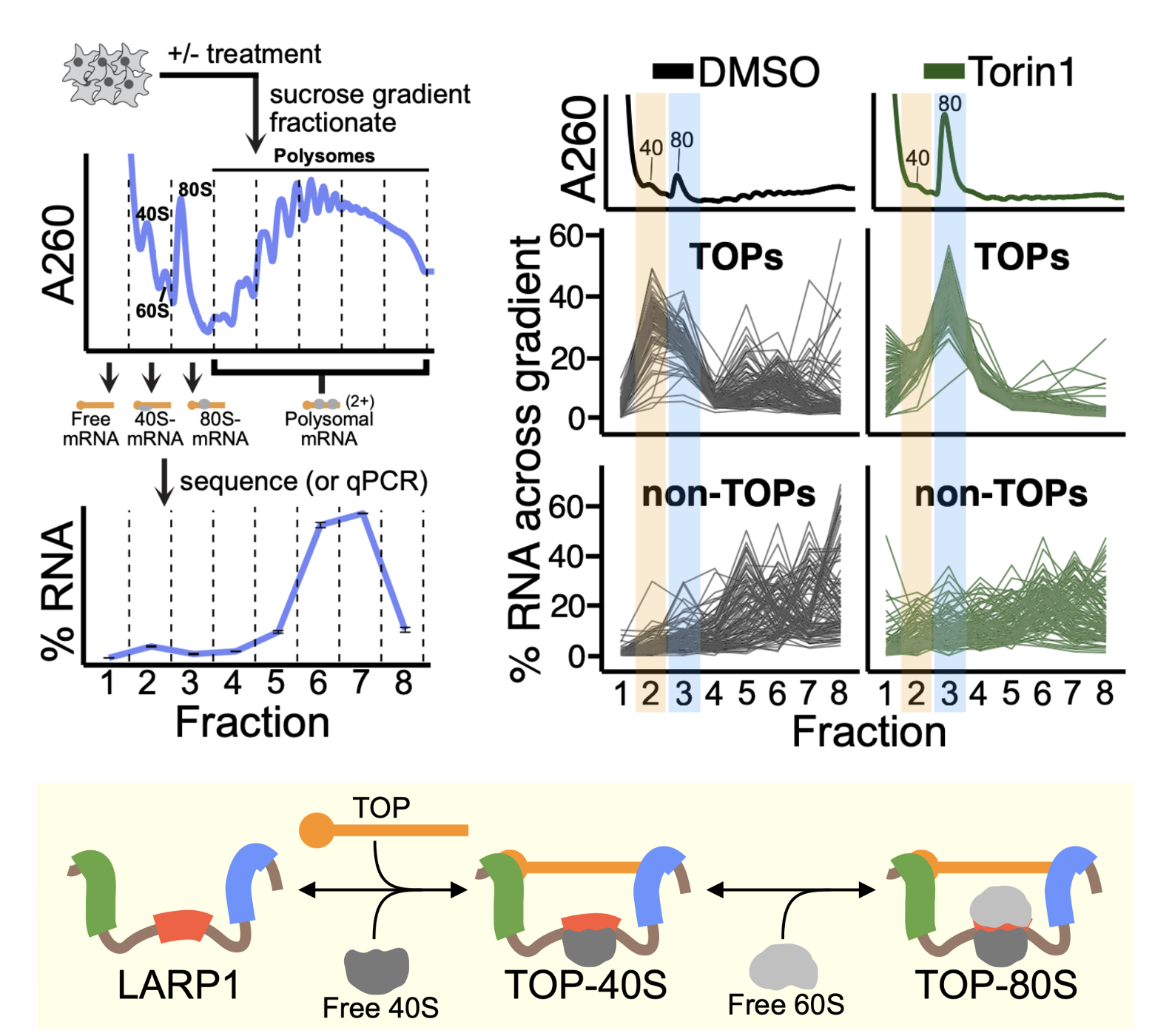
We recently have identified direct connections between ribosome collisions and cellular signaling pathways and we are exploring these connections at the molecular level. Ongoing work in this area is defining the interactions of ribosome-mediated quality control and cellular signaling pathways in tissues where protein synthesis regimes may be different (skin, cancer cells, neurons). Our interests in ribosome recycling and rescue have raised our interest in ribosome homeostasis and how it contributes to the regulation of gene expression. Why do ribosome deficiencies in different tissues lead to differential sensitivities to these deficiencies? How does the cell balance the demands of protein synthesis with the regulation of initiation, elongation, termination, and recycling steps of this process to ensure that gene expression happens appropriately in order to sustain the life of the organism? We also continue to be interested in connections between ribosome read-through at stop codons and nonsense-mediated decay, based in part on the relevance of such models to therapeutic treatment for diseases originating from premature stop codons in critical genes.